AI under the microscope: the algorithms powering the search for cells

Deep learning is driving the rapid evolution of algorithms that can automatically find and trace cells in a wide range of microscopy experiments
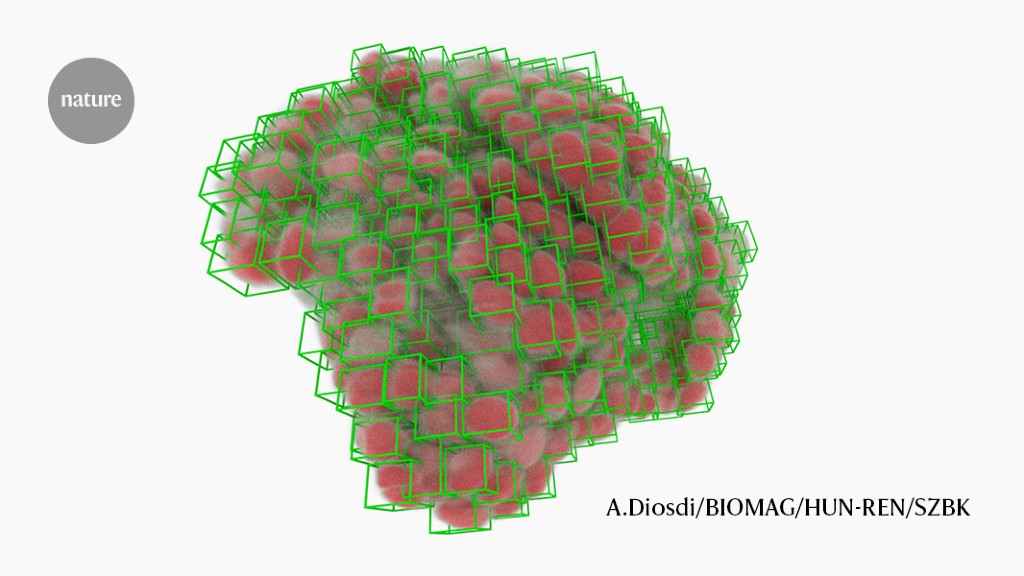
Cancer-cell nuclei (green boxes) picked out by software using deep learning.Credit: A. Diosdi - imaging; T. Toth & F. Kovacs - analysis; A. Kriston - visualization/BIOMAG/HUN-REN/SZBK
Any biology student can pick a neuron out of a photograph. Training a computer to do the same thing is much harder. Jan Funke, a computational biologist at the Howard Hughes Medical Institute’s Janelia Research Campus in Ashburn, Virginia, recalls his first attempt 14 years ago. “I was arrogant, and I was thinking, ‘it can’t be too hard to write an algorithm that does it for us’,” he says. “Boy, was I wrong.”
People learn early in life how to ‘segment’ visual information — distinguishing individual objects even when they happen to be crowded together or overlapping. But our brains have evolved to excel at this skill over millions of years, says Anna Kreshuk, a computer scientist at the European Molecular Biology Laboratory in Heidelberg, Germany; algorithms must learn it from first principles. “Mimicking human vision is very hard,” she says.
But in life-science research, it’s increasingly required. As the scale and complexity of biological imaging experiments has grown, so too has the need for computational tools that can segment cellular and subcellular features with minimal human intervention. This is a big ask. Biological objects can assume a dizzying array of shapes, and be imaged in myriad ways. As a result, says David Van Valen, a systems biologist at the California Institute of Technology in Pasadena, it can take much longer to analyse a data set than to collect it. Until quite recently, he says, his colleagues might collect a data set in one month, “and then spend the next six months fixing the mistakes of existing segmentation algorithms”.
The good news is that the tide is turning, particularly as computational biologists tap into the algorithmic architectures known as deep learning, unlocking capabilities that drastically accelerate the process. “I think segmentation overall will be solved within the foreseeable future,” Kreshuk says. But the field must also find ways to extend these methods to accommodate the unstoppable evolution of cutting-edge imaging techniques.
A learning experience
The early days of computer-assisted segmentation required considerable hand-holding by biologists. For each experiment, researchers would have to carefully customize their algorithms so that they could identify the boundaries between cells in a particular specimen.
Image-analysis tools such as CellProfiler, developed by imaging scientist Anne Carpenter and computer scientist Thouis Jones at the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and ilastik, developed by Kreshuk and her colleagues, simplify this process with machine learning. Users educate their software by example, marking up demonstration images and creating precedents for the software to follow. “You just point on the images and say, this is what I want,” Kreshuk explains. That strategy is limited in its generalizability, however, because each training process is optimized for a particular experiment — for example, detecting mouse liver cells labelled with a specific fluorescent dye.
Deep learning produced a seismic shift in this regard. The term describes algorithms that use neural network architectures that loosely mimic the organization of the brain, and that can extrapolate sophisticated patterns after training with large volumes of information. Applied to image data, these algorithms can derive a more robust and consistent definition of the features that represent cells and other biological objects — not just in a given set of images but across multiple contexts.
A deep learning framework known as U-Net, developed in 2015 by Olaf Ronneberger, a computer scientist at the University of Freiburg in Germany, and his colleagues, has proved particularly transformative. Indeed, it remains the underlying architecture behind most segmentation tools almost a decade later.
Many initial efforts in this space focused on identifying cell nuclei. These are large and ovoid, with little variation in appearance across cell types, and virtually every mammalian cell contains one. But they can still pose a challenge in cell-dense tissue samples, says Martin Weigert, a bioimaging specialist at the Swiss Federal Institute of Technology in Lausanne. “You can have very tightly packed nuclei. You don’t want a segmentation method to just say it’s a gigantic blob,” he says. In 2019, a team led by Peter Horvath, an imaging specialist at the Biological Research Centre in Szeged, Hungary, used U-Net to develop an algorithm called nucleAIzer. It performed better than hundreds of other tools that had competed in a ‘Data Science Bowl’ challenge for nuclear segmentation in light microscopy the year before1.
Even if software can find the nucleus, it remains tricky to extrapolate the shape of the rest of the cell. Other algorithms aim for a more holistic strategy. For example, StarDist, developed by Weigert and his collaborator Uwe Schmidt, generates star-shaped polygons that can be used to segment nuclei while also extrapolating the more complex shape of the surrounding cytoplasm.

Cells segmented by the CellPose software (left) and the ‘flow fields’ that it used to do the task (right).Credit: Carsen Stringer & Cell Image Library (CC-BY)
CellPose takes a more generalist approach. Developed by Marius Pachitariu, a computational neuroscientist at Janelia, and co-principal investigator Carsen Stringer in 2020, the software derives ‘flow fields’ that describe the intracellular diffusion of the molecular labels commonly used in light microscopy. “We came up with this representation of cells that’s basically described by these vector dynamics, with these flow fields kind of pushing all the pixels towards the centre of the cell,” says Pachitariu. This allows CellPose to confidently assign each pixel in a given image to one cell or another with high accuracy — and more importantly, with broad applicability across light microscopy methods and sample types.
“One of the magic things about CellPose was that … even with cells that are touching, it can split them apart just fine,” says Beth Cimini, a bioimaging specialist at the Broad who currently runs the CellProfiler project.
Basic training
These steady gains in deep-learning-based segmentation stem only partly from advances in algorithm design — most methods still riff on the same underlying U-Net foundation.
Instead, the key determinant of success is training. “Better data, better labels — that’s the secret,” says Van Valen, who led the development of a popular segmentation tool, called DeepCell. Labelling entails assembling a collection of microscopy images, delineating the nuclei and membranes and other structures of interest, and feeding those annotations into the software so that it can learn the features that define those elements. For CellPose, Pachitariu and Stringer spent half a year collecting and curating as many microscopy images as they could to build a large and broadly representative training data set — including non-cell images to provide clear counterexamples.
But building a vast, manually annotated training set can quickly become overwhelming, and deep-learning experts are developing strategies to work smarter rather than harder.
Diversity is one priority. “Having a few of a lot of different things is better than having very much of the same,” says Weigert. For example, a collection of images of brain, muscle and liver tissue with a variety of staining and labelling approaches is more likely than are images of just one tissue type to produce results that generalize across experiments. Horvath also sees value in including imperfections — for example, out-of-focus images — that teach the algorithm to overcome such problems in real data.
Another increasingly popular strategy is to let algorithms do bulk annotation and then bring humans in to fact-check. Van Valen and his colleagues used this ‘human in the loop’ approach to develop the TissueNet image data set, which contains more than one million annotated cell-nucleus pairs. They tasked a crowdsourced community of novices and experts with correcting predictions by a deep-learning model that was trained on just 80 manually annotated images. Van Valen’s team subsequently developed a segmentation algorithm called Mesmer, and showed that this could match human segmentation performance after training it with TissueNet data22. Pachitariu and Stringer likewise used this human-in-the-loop approach to retrain CellPose to achieve better performance on specific data sets3, using as few as 100 cells. “On a new data set, we think a user can probably train their model in the loop within an hour or so,” Pachitariu says.
Even so, retraining for new tasks can be a chore. To streamline the process, Kreshuk and Florian Jug, a computational biologist at the Human Technopole Foundation in Milan, Italy, have created the BioImage Model Zoo, a community repository of pre-trained deep-learning models. Users can search it to find a ready-to-use model for segmenting images, rather than struggling to train their own. “We are trying to make the zoo really usable by biologists who are not very deep into deep learning,” says Kreshuk.
Indeed, the unfamiliarity of many wet-lab scientists with the intricacies of deep-learning algorithms is a broad roadblock to deployment, says Cimini. But there are many avenues to accessibility. For example, she attributes CellPose’s success to its straightforward graphical user interface as well as its segmentation capabilities. “Putting the effort in to make stuff very friendly and approachable and non-scary is how you reach the majority of biologists,” she says. Many algorithms, including CellPose, StarDist and nucleAIzer, are also available as plug-ins for popular image-analysis tools including ImageJ/Fiji, napari (Nature 600, 347–348; 2021) and CellProfiler.
Pushing the boundaries
Just a few years ago, Cimini says, she never imagined such rapid progress. “At least for nuclei — and probably even for cells, too — we’re actually getting to the point that within a few years, this is going to be a solved problem.”
Researchers are also making headway with more-challenging types of images. For example, many spatial transcriptomics studies entail multiple rounds of tissue labelling and imaging, in which each label or collection of labels reveals the RNA transcripts for a specific gene. These are then reconstructed alongside images of the cells themselves to create tissue-wide gene-expression profiles with cellular resolution.
But identification and interpretation of gene-expression ‘spots’ is tough to automate. “When you actually open these raw images, you’ll see that there are just far too many spots for human beings to ever manually label,” says Van Valen. This, in turn, makes training difficult. Van Valen’s team has developed a deep-learning network that can confidently discern these spots with help from a classical computer-vision algorithm4. The researchers then integrated this network into a larger pipeline called Polaris, a generalizable solution that can be applied for end-to-end analysis of a wide range of spatial transcriptomics experiments.
By contrast, analysis of 3D volumes for light microscopy remains stubbornly difficult. Weigert says there is a profound shortage of publicly available 3D imaging data, and it is a chore to make such data useful for algorithmic training. “Annotating 3D data is such a pain — this is where people go to cry,” says Weigert. The variability in data quality and formats is also more extreme than for 2D microscopy, Pachitariu notes, necessitating larger and more-complicated training data sets.
There has, however, been remarkable progress in segmentation of 3D data generated by ‘volume electron microscopy’ methods. But interpreting electron micrographs throws up fresh challenges. “Whereas in light microscopy you have to learn what is signal and what is background, in electron microscopy you have to learn to distinguish what makes your signal different from all the other kinds of signal,” says Kreshuk. Volume electron microscopy heightens this challenge, requiring reconstruction of a series of thin sample sections that document cells and their environments with remarkable detail and resolution.
These capabilities are particularly important in connectomics studies that seek to generate neuronal ‘wiring maps’ of the brain. Here, the stakes are especially high in terms of accuracy. “If we make, on average, one mistake per micron of a neural fibre or per axon length, then the whole thing is useless,” says Funke. “This is one of the hardest problems you can have.” But the vast volumes of data also mean that algorithms need to be efficient to complete reconstruction in a reasonable time frame.
As with light microscopy, U-Net has delivered major dividends. In a preprint posted in June, the FlyWire Consortium — of which Funke is a member — described applying a U-Net-based algorithm to reconstruct the wiring of an adult fly brain, comprising roughly 130,000 neurons5. An assessment of 826 randomly chosen neurons found that the algorithm achieves 99.2% accuracy relative to human evaluators. Segmentation algorithms for connectomics are now largely mature, Funke says — although fact-checking these circuit diagrams at whole-brain scale remains a daunting task. “The part that we’re nervous about now is, how do we proofread?”
A singular solution
Interoperability across imaging platforms also remains a challenge. An algorithm trained on samples labelled with the haematoxylin and eosin stains commonly used in histology might not perform well on confocal microscopy images, for example. Similarly, methods designed for segmentation in electron microscopy are generally incompatible with light microscopy data.
“For each technology, they capture biological specimens at significantly varying scales and emphasize distinct features due to the different staining, different treatment protocols,” says Bo Wang, an artificial-intelligence specialist at the University of Toronto in Canada. “So, when you think about designing or training a deep-learning model that works across the full spectrum of these methods, that means really simultaneously excelling in different tasks.”
Wang is bullish about ‘foundation models’ that can truly generalize across imaging data formats, and helped to coordinate a data challenge at last year’s Conference on Neural Information Processing Systems (NeurIPS) for various groups to test their mettle in developing solutions.
In addition to bigger and broader training sets, such models will almost certainly require computational architectures beyond the comfortable confines of U-Net. Wang is enthusiastic about transformers — algorithmic tools that make it easier for deep learning to discern subtle but important patterns in data. Transformers are a central component of both the large language model ChatGPT and the protein-structure-predicting algorithm AlphaFold, and the winning algorithm at the 2022 NeurIPS challenge leveraged transformers to achieve a decisive advantage over other approaches. “This helps the model focus on relevant cellular or tissue structures while ignoring some of the noise,” Wang says. Numerous groups are now developing foundation models; this month, for example, Van Valen and his team posted a preprint describing their CellSAM algorithm6. Wang is optimistic that first-generation solutions will emerge in the next few years.
In the meantime, many researchers are moving on to more-interesting applications of the tools they’ve developed. For example, Funke is using segmentation-derived insights to classify the functional characteristics of neurons based on their morphology — discerning features of inhibitory versus excitatory cells in connectomic maps, for instance. And Horvath’s team collaborated on a method called deep visual proteomics, which leverages structural and functional insights from deep-learning algorithms to delineate specific cells in tissue samples that can then be precisely plucked out and subjected to deep transcriptomic and proteomic analysis7. This could offer a powerful tool for profiling the molecular pathology of cancers and identifying appropriate avenues for treatment.
These prospects excite Kreshuk as well. “I hope in the near future we can make morphology space really quantitative,” she says, “and analyse it together with the omics space and see how we can mix and match this stuff.”
Nature 623, 1095-1097 (2023)
doi: https://doi.org/10.1038/d41586-023-03722-y
This story originally appeared on: Nature - Author:Michael Eisenstein

















